2. Water Sorption on Sodium Faujasite by Monte Carlo Simulations
Faujasitic zeolites are employed in a range of industrial contexts including separation processes, gas purification and dehydration, and shape selective catalysis. The simulation of the interaction of small molecules with faujasite based zeolites is increasingly practical and the resulting information provides the basis for efficient materials selection and design. When experimental data are scarce and empirical models insufficiently accurate, the MedeA®[1] Environment can be employed to provide quantitative property data for faujasitic zeolite systems.
Keywords: sorption, Grand Canonical Monte Carlo, GCMC, faujasite, water
2.1. Introduction
Zeolites are crystalline microporous silico-aluminates that may be synthesized in hydrothermal conditions to produce industrial adsorbents of controlled pore geometry. The presence of aluminium in the framework causes charge defects that are compensated by extra-framework cations such as Na, K, Ba, etc. [2]. Water plays an important role in the sorption properties of zeolites.
Understanding physisorption in these materials requires a molecular-level approach and an appropriate account of the various types of interaction, especially when polar components like water are considered. Grand Canonical Monte Carlo simulation [3] is a well-known technique to address sorption on such systems, as it is applicable when molecules are densely packed in the nanopores with strong interactions with the cations. Here MedeA GIBBS, the Monte Carlo module of the MedeA software, is used and comparison is made with available experimental data.
2.2. Molecular Simulation
The number of sorbed molecules in a microporous solid may be obtained by a simulation in the Grand Canonical ensemble [3], where the imposed parameters are the chemical potentials \(\mu\)iof the adsorbed compounds, temperature T and volume V. The adsorbent is considered as rigid, i.e. the deformation energy of the framework is neglected. In sorption studies, it is convenient to impose partial pressures Pi, which are measured by pressure sensors in experiments, instead of chemical potentials \(\mu\)i. At pressures below 1 bar, both variables are related by:
Where \(\mu\)i0 is the chemical potential at a reference pressure \(P_{i0}\).
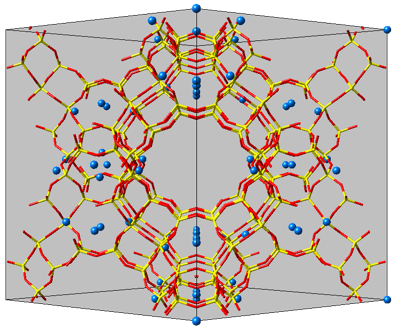
Figure 2.2.1 Unit cell of the Sodium-Faujasite Na56Y model used in the Monte Carlo simulations. The unit cell contains 56 sodium cations.
2.3. Computational Details
A faujasite structure (Figure 2.2.1) can be introduced and used in MedeA from any personal, commercial or open-access library [4] [5]. The sodium cations are located according to previous studies [2] [6], and forcefield parameters are assigned using a combination of Lennard-Jones and electrostatic charges [6] [7]. Periodic boundary conditions are applied.
The potential energy of interaction between the sorbed molecules and the adsorbent is pre-calculated over a finely meshed grid that is overlaid on the simulation unit-cell. In this approach, the framework atoms have no freedom of movement, which accelerates the calculation.
A Grand Canonical Monte Carlo (GCMC) simulation consists in inserting and deleting molecules from the nanopores and exploring all possible ways that they can pack in the pores. The method involves thus elementary Monte Carlo “moves” of insertion, deletion, rotations and translations of water molecules. The rules to perform some changes and reject others are such that the equilibrium conditions at the desired pressure and temperature are satisfied [3]. For a given set of conditions, the outcome of a Monte Carlo simulation is not a final configuration of the water molecules in the unit cell, but an ensemble of representative configurations. From this ensemble, average properties can be computed to compare with experimental measurements. For instance, the average number of molecules may be compared with the equilibrium loading that is recorded by a thermogravimetry. Repeating the calculation for different temperatures in a loop allows to compute water desorption when heating at constant pressure (Figure 2.3.1).
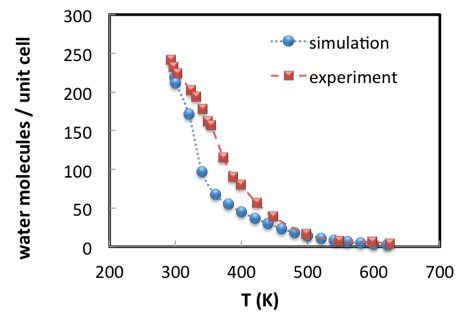
Figure 2.3.1 Water sorption in Na56Y faujasite versus temperatures. Water partial pressure is 1,690 Pa.
The calculation of an adsorption isotherm is performed in an automated loop over pressures (Figure 2.3.2).
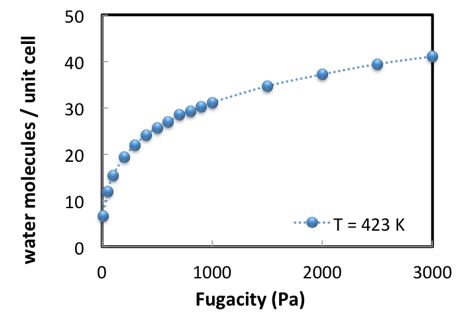
Figure 2.3.2 Water sorption in Na56Y at 423 K; simulation results.
2.4. Results
The good agreement between simulation results and experimental data (Figure 2.3.1) illustrates the predictive capacity of Monte Carlo simulation for zeolites with well-designed forcefields [7] [8]. Simulated results are determined by the accurate description of electrostatic and interatomic interactions [9] and considerable effort has been dedicated to providing access to state-of-the-art forcefields for sorption simulation within the MedeA Environment. In addition to supporting leading scientific developments, MedeA GIBBS allows the automation of calculations, such that an adsorption isotherm can be obtained through multiple GCMC simulations administered from a single controlling computation, for example. MedeA GIBBS also provides post-processing tools to visualize molecules in nanopores, characterize their average position, draw the adsorption isotherms, and so on. Computations on systems such as the ones described in this note are completed in only a few hours on a few cpus.
With MedeA GIBBS, Monte Carlo simulation can now be efficiently employed in the simulation of a range of system types and compositions. The resulting capability makes sorption simulation, using GCMC, accessible in optimizing and understanding separation, ion exchange, carbon sequestration, shape selectivity, fluid catalytic cracking, and pressure swing adsorption based processes.
- download: