7. Stability of Alkaline-Earth Hydrides
Hydrides containing alkaline-earth metals are prototypes for hydrogen storage materials. For this purpose the heat of formation and the mechanical properties are of fundamental interest. First-principles electronic structure calculations provide systematic values for these materials properties. For the heat of formation the agreement of computed and experimental data is good. Presently, no experimental data for the elastic coefficients of these metal hydrides are available thus leaving the computed data as the sole source.
Keywords: hydrogen storage materials, alkaline earth hydrides, heat of formation, elastic coefficients, computation
7.1. Technological context
In the context of e-mobility, metal hydrides are a promising option for on-board storage of hydrogen. One of the fundamental properties of a hydrogen storage material is the energy released/required for the uptake and discharge of hydrogen. In addition, high capacity, light weight, fast charge/discharge, mechanical stability, safety, low cost, and environmental compatibility are required for a successful material. Alkaline-earth metal hydrides such as MgH2 are prototypical materials serving as reference for the design of more suitable and more complex materials.
7.2. Computed materials properties
The computed heat of formation of the alkaline earth hydrides describes very well trends of the binding energy as a function of atomic number as illustrated in (Figure 7.2.1). The two levels of theory, called local density approximation (LDA) and the generalized gradient approximation (GGA) bracket the experimental data. The deviation between different experimental data is of similar magnitude as that between the computed GGA results and the experimental data (see, for example CaH2 and SrH2 in (Figure 7.2.1)).

Figure 7.2.1 Computed (LDA and GGA) and experimental heat of formation of alkaline-earth hydrides. After Ref. [2]
The computed bulk modulus of this series of hydrides shows a maximum for CaH2 with a decrease for heavier metal atoms (see Figure 7.2.2)).
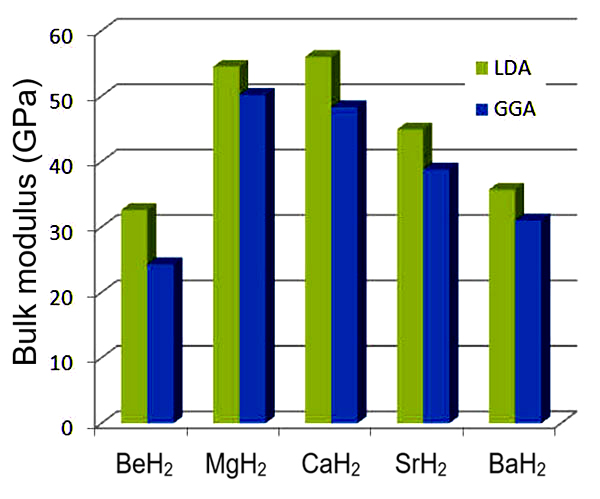
Figure 7.2.2 Computed bulk modulus of alkaline-earth hydrides.
MgH2 ranks second. Somewhat surprisingly BeH2 has the lowest bulk modulus in this series. Systematically the values obtained at the LDA level of theory is higher than those computed with GGA. This is consistent with the results for the heats of formation. The LDA tends to give bond energies which are too strong compared with experiment, while GGA predicts binding energies, which are often too small.
7.3. Significance
The accuracy of the computed heats of formation of the alkaline-earth hydrides serves as a solid reference to explore a wide range of materials design options. In most cases it is much faster and more cost effective to compute a heat of formation than to synthesize and characterize a new compound and then to perform calorimetric experiments, which may be hindered by slow kinetics.
The accurate experimental determination of fundamental mechanical properties such as the bulk modulus or the shear modules is quite difficult while today’s level of first-principles computations provide values which are comparable in accuracy and reliability with experiment. Thus, computations offer a remarkably efficient source to obtain this property.
MedeA modules used in this application
MedeA®[1] Environment
MedeA Pearson
MedeA VASP
MedeA MT
MedeA Phonon
- download: