16. Atomic Structure of Hydrodesulfurization (HDS) Catalysts
Keywords: catalysts, surfaces, HDS, Co(Ni)MoS, first-principles, computations
First-principles calculations reveal the atomistic structures of the active phases of CoMoS and NiMoS hydrodesulfurization catalysts. The reliable determination of the catalyst surface is critical, as it represents the starting point for subsequent adsorption and reaction path simulations. The predicted dominant structures are consistent with experimental STM, EXAFS and magnetic susceptibility measurements.
16.1. Background
Increased environmental concerns made the production of “greener” fuels a priority for the petroleum industry. To accomplish this goal the discovery and development of catalysts were needed for processes such as sulfur reduction. Hydrodesulfurization (HDS) is the removal of sulfur, under hydrogen pressure from natural gas and other petroleum products. CoMoS and NiCoS are widely used catalysts for HDS. Experimental characterization indicates that the active phases consist of MoS2 layers decorated by promoter Co/Ni atoms at the edges of the catalyst. However, the specific atomic details were unknown. First-principles calculations were carried out to determine the atomic structure of the CoMoS and NiMoS catalysts as a function of reaction conditions to identify the structure of the active phases at typical HDS conditions [1], [2].
16.2. Computed Results
The MoS edge energy diagrams are shown in Figure 16.2.1 for the metal and sulfur edges (M- and S-edge) as a function of reaction conditions. Reactions conditions are determined by the partial pressures of H2S and H2(p(H2S)/p(H2). Typical reaction conditions for the HDS of FCC gasoline occur at T= 525 K, indicated as the red vertical lines. As shown, the preferred edge differs for the 100% decorated Co- and Ni- promoted MoS catalysts: the S- and M-edge have the lowest energies for CoMoS and NiMoS, respectively. At HDS conditions, the 50% decorated M-edge for CoMoS is computed to be isoenergetic with the 100% S-edge structure, whereas for NiMoS the 50% decorated M-edge is isoenergetic with the 100% M-edge structure. These predictions are in line with experimental measurements. For example, for CoMoS the 50% coverage structure is compatible with magnetic susceptibility measurements by Okamoto [3] and the calculated 100% decorated structure is consistent with STM [4] and EXAFS data [5]. Similarly, the simulated NiMoS structures are in agreement with EXAFS reports [6].
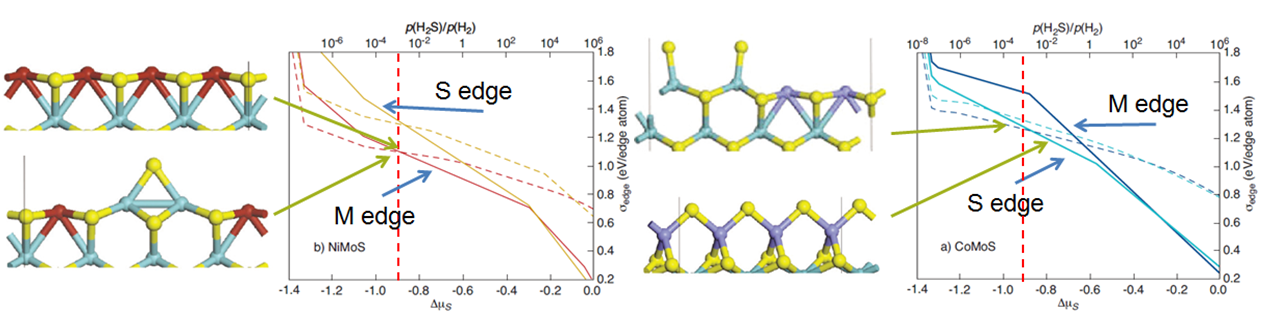
Figure 16.2.1 Edge energy diagrams showing the values for the M- and S-edges of Ni-MoS and Co-MoS (left and right, respectively) as a function of reaction conditions. Red vertical lines indicate HDS reaction conditions. Solid and dashed lines denote 100% and 50% Ni/Co coverage, respectively. Dominant atomic structures at typical HDS conditions are shown (adapted from [1]).
16.3. Conclusion
The atomic structure of catalytic surfaces is a defining characteristic determining the catalytic activity by controlling the strength of steric, electrostatic, and orbital interactions with reactants. The starting point for the analysis and optimization of existing catalyst systems is the geometry and composition of the chemically active phase. Given the importance of developing clean fuels, an in-depth understanding of Co(Ni)MoS catalyst structure is a critical step in the development of highly effective HDS catalysts.
First-principles calculations are a reliable source for predicting the atomistic structure, stability, and intrinsic chacteristics for catalytic surfaces as demonstrated here for the case of Co/Ni promoted MoS catalysts. With the rapidly increasing performance of computational resources, the fundamental properties controlling the catalytic activity for increasingly complex systems such as nanostructures can be predicted with high confidence, thus guiding the design of highly efficient and selective industrial catalysts.
MedeA modules used in this application
MedeA Environment
MedeA VASP
- download: