17. Catalytic Isomerization of pent-2-ene in H-ZSM-22
Keywords: catalysts, isomerization, ZSM-22, zeolites, acidity, first-principles, computations
First-principles calculations using VASP reveal the lowest energy reaction pathway for the catalyzed skeletal isomerization of pent-2-ene by the acidic zeolite H-ZSM-22. Three potential mechanisms were evaluated: an ethyl-shift pathway, a dimethylcyclopropane (DMCP) intermediate pathway, and a pathway involving an edge-protonated DMCP species. The results indicate that the DMCP intermediate pathway is the kinetically preferred pathway with a classical barrier height of 98 kJ mol-1. Evident in the calculations is the influence of the transient intermediate stability along the reaction path, with secondary carbenium ions leading to energetically favored mechanisms.
17.1. Background
A key process in the petrochemical refining industry is the isomerization of hydrocarbons, usually carried out using bi-functional catalysts. [1] These catalysts consist of noble metal particles dispersed over an acidic zeolite: the metal and zeolite accelerate dehydrogenation-hydrogenation and isomerization reactions, respectively. The isomerization step involves the skeletal rearrangement of an alkene carbon backbone into a branched isomer. Experimentally, the alkene restructuring is the rate-determining step in overall hydrocarbon isomerization. An in-depth understanding of the chemical mechanism and atomic details are needed to enable the rationalization of observed reactivities and inform efforts at developing more effective catalytic frameworks for hydrocarbon refinement. First-principles calculations were carried out to determine the underlying chemical mechanism for the isomerization of pent-2-ene in H-ZSM-22. [2]
17.2. Computed Results
The initial reaction of pent-2-ene within the H-ZSM-22 framework is chemisorption at the zeolite acid site, producing an alkoxide intermediate. The activation energy for this step was calculated to be thermally accessible (52 kJ mol-1). Three different mechanisms for the subsequent C5 isomerization were considered, namely, 1) the ethyl-shift pathway, 2) the edge- protonated DMCP transition state patwhay, and 3) the dimethylcyclopropane (DMCP) intermediate pathway. The latter is illustrated in Figure 17.2.1. Comparison of the activation energies for the overall rate-determining step indicates that the DMCP intermediate pathway is the kinetically preferred C5 isomerization mechanism.
As illustrated in Figure 17.2.1 (right), the largest energy barrier along that pathway is computed to be 98 kJ mol-1. The other pathways have overall energy barriers larger by 82 and 12 kJ mol-1 for the mechanisms through 1) and 3)(Eact=110kJ/mol in Figure 17.2.1 (right)), respectively. The energetics resolving the pentene isomerization pathways can be attributed to the relative stability of the carbenium ion involved in the transition state structures. Tertiary or secondary carbocations are more stable than primary carbocations. The DMCP intermediate pathway involves a secondary carbenium ion, whereas the ethyl-shift pathway requires primary carbenium formation.
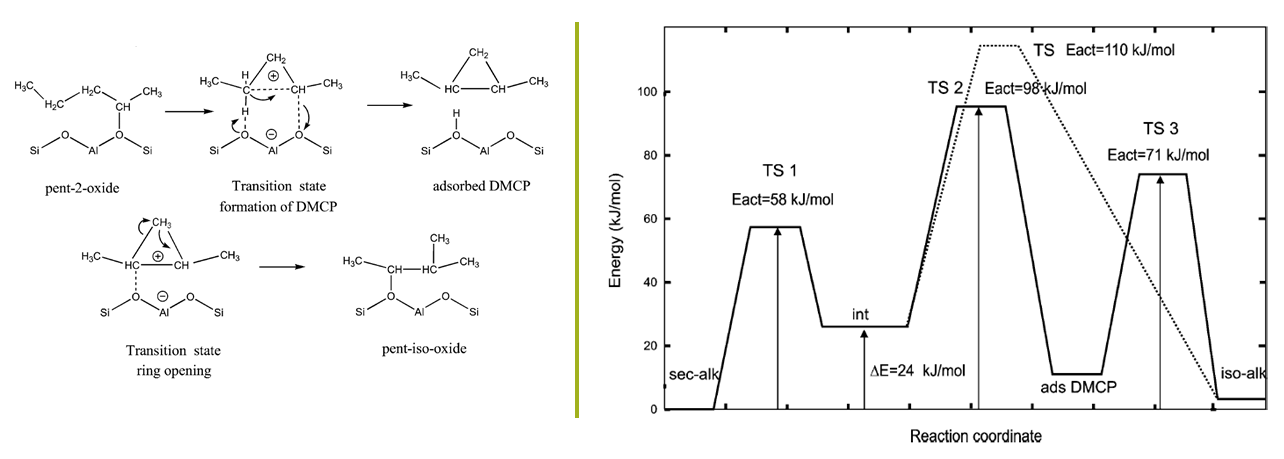
Figure 17.2.1 The minimum energy C5 isomerization pathway in the acidic H-ZSM-22 zeolite proceeding via a dimethylcyclopropane (DMCP) intermediate: (Left) DMCP Intermediate pathway mechanism, and (Right) DMCP Intermediate pathway energy profile (solid line) along with that for the edge-protonated DMCP pathway, for comparison (dotted line). (Adapted from [2])
17.3. Significance
Determining the chemical mechanisms underlying complex catalytic processes is key in understanding the underlying factors controlling the overall reaction kinetics. Knowledge of the preferred elementary reaction pathways enables the search for new catalytic systems that exhibit greater activity and selectivity by varying the sterics, electrostatics, and acid/base characteristics through changing structure and composition. The chemical mechanisms and details for catalysis of C5 isomerization in H-ZSM-22 are of wide interest, given the importance of hydrocarbon isomerization to the petrochemical industry.
First-principles calculations are a reliable source for predicting the atomistic structure, chemical mechanisms, and activation energies for complex catalytic processes as demonstrated here for the case of zeolite catalyzed pentene isomerization. With the rapidly increasing performance of computational resources, the multi-step reaction paths and rate-limiting energies characterizing the catalytic activity for increasingly complex systems such as nanostructures can be predicted with high confidence, thus guiding the design of highly efficient and selective industrial catalysts.
17.4. Methods
The present calculations were performed using the Vienna Ab Initio Simulation Package (VASP) and details can be found in [2].
- download: