37. Organic Materials Properties: Densities, Cohesive Energies, and Heats of Vaporization
This application note describes the calculation of densities, cohesive energy densities (CEDs), and enthalpies of vaporization for a range of straight chain hydrocarbon fluids. The construction, simulation, and analysis methodologies employed are reviewed; and the accuracy of atomistic simulation for organic materials and polymers illustrated. Mean absolute errors are 0.29% and 0.17% for densities and heats of vaporization respectively for the PCFF+ forcefield.
Keywords: MedeA environment, LAMMPS, cohesive energy density (CED), hydrocarbons, density, forcefield, PCFF+, molecular dynamics (MD)
37.1. Purpose
Organic materials in their various forms are critically important technologically. Hence there is great interest in the accurate simulation of their properties. Here we provide an illustration of the simulation of organic fluids, employing straight chain hydrocarbon systems as the basis for the study. These simple hydrocarbon systems are of importance, given their uses as fuels and solvents, and their properties are well characterized experimentally, providing a sound basis for methodological validation.
The methods employed include model construction, making use of the MedeA ®[1] Polymer Builder, molecular dynamics calculation, using MedeA LAMMPS, and detailed analysis of simulation output managed by the MedeA JobServer.
Atomistic molecular dynamics calculations rely on rapidly calculated forces on component atoms followed by integration of Newtonian equations of motion to yield a comprehensive description of the behavior of a given system as a function of time. The combinations of functional form and parameters, required to calculate system energies and forces, are collectively termed forcefields [2], and forcefield accuracy governs the quality of simulated properties.

Figure 37.1.1 A snapshot from a molecular dynamics calculation for n-hexane at 25 oC and 1 atm.
Experience with the simulation techniques illustrated here indicates that the simulation accuracies obtained for hydrocarbons are representative of those achievable for arbitrary organic systems. Modern forcefields for organic systems have been derived on the basis of broad coverage of common molecular components. Where forcefield coverage is available, simulations may be undertaken and high levels of accuracy obtained in advance of experiment [3], [4], [5], [6], and [7]. Forcefields for organic systems are constructed to be broadly applicable, and as the current example illustrates, the agreement between simulation and experiment is generally excellent, with simulated properties typically within a percentage point of observed values.
As noted, the simulations described here employ LAMMPS [8], the Sandia National Laboratories ‘Large-scale Atomic/Molecular Massively Parallel Simulator’, which yields highly optimized molecular dynamics performance, particularly when coupled with multiprocessor compute servers. The MedeA framework provides a flexible environment for using LAMMPS which allows facile model construction, forcefield assignment, simulation workflow construction, calculation management, and analysis.
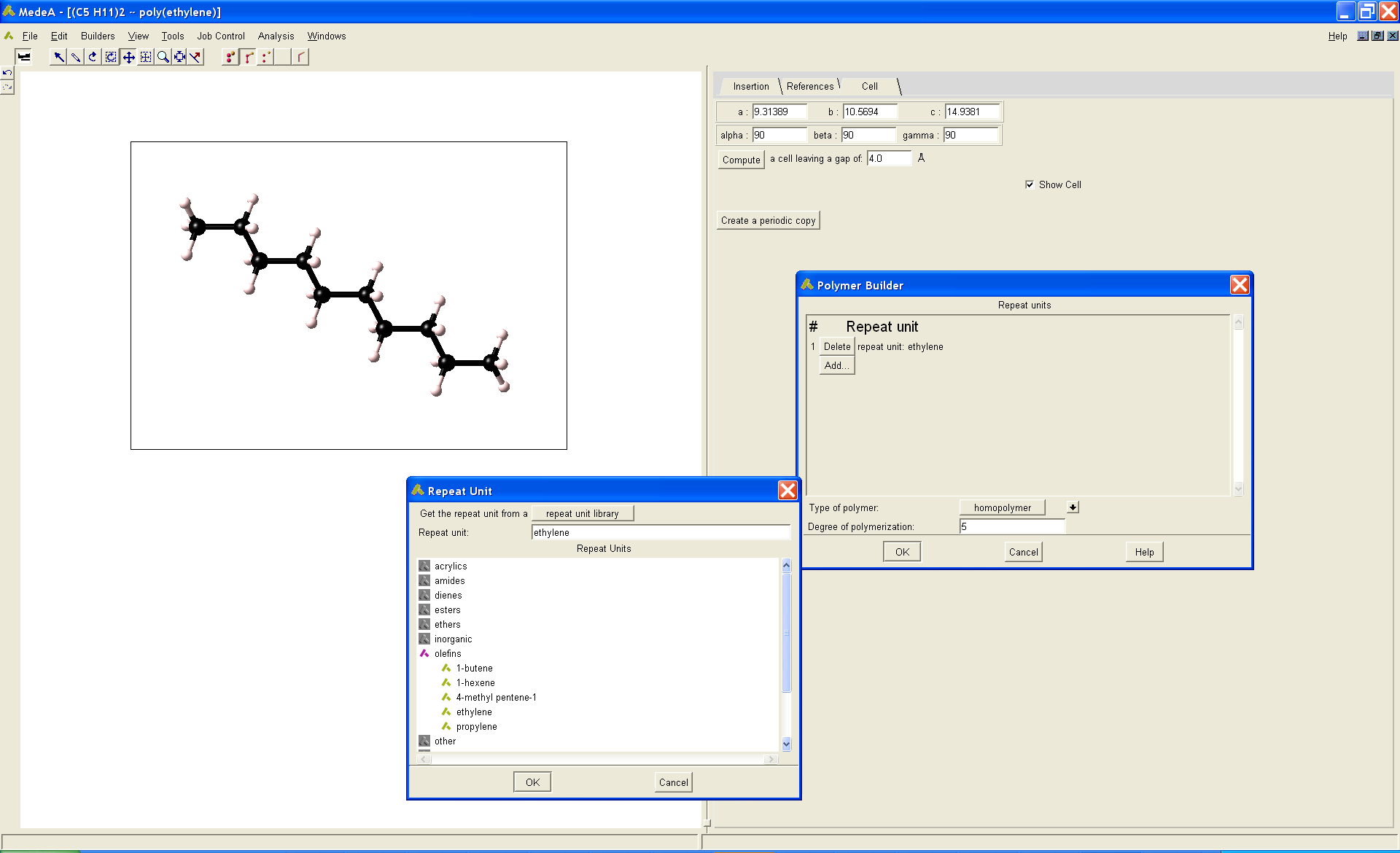
Figure 37.1.2 The MedeA Polymer Builder allows the rapid construction of a comprehensive range of oligomers and polymers.
37.2. Building Hydrocarbon Systems
Atomistic molecular dynamics simulations require a starting model. Here initial hydrocarbon models were constructed using the MedeA Polymer Builder, as illustrated in (Figure 37.1.2), where n-decane is treated as a 5-residue polyethylene oligomer. The MedeA Polymer Builder constructs polymer models from a library of standard repeat units, and provides a comprehensive treatment of composition and stereochemistry. If required, the repeat unit library can be readily extended by the user, such that any desired chain-like system can be constructed and simulated. The hydrocarbon models described here represent examples of the methodologies typically applied using the MedeA Environment and LAMMPS to model polymeric system.
37.3. Creating Simulation Models
The simulation of the physical and energetic properties for a fluid requires a condensed model. Such a model may be readily constructed based on the isolated molecular description of (Figure 37.1.2). The MedeA framework supports a variety of tools which can be used to create compact models. For example, the application of a translational symmetry rapidly leads to a ‘supercell’ model. This model can in turn be compacted to typical liquid density using a molecular dynamics simulation protocol which employs a progressively reduced external applied pressure, as shown schematically in flowchart form in (Figure 37.3.1). Liquid models, and indeed condensed polymer models, can also be readily created using the MedeA Amorphous Materials Builder [9].
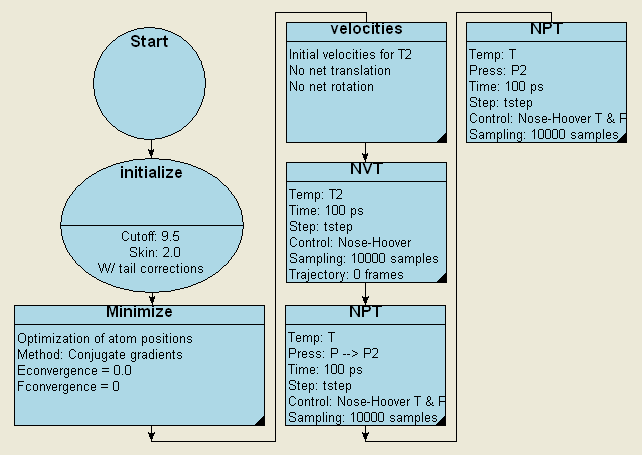
Figure 37.3.1 An illustrative flowchart for the MedeA LAMMPS based creation of condensed hydrocarbon models. The first NPT stage employs an external pressure which is reduced from a high value to 1 atmosphere in a linear ramp over 100ps. The second NPT molecular dynamics stage is computed at a constant 1 atmosphere pressure.
37.4. Forcefield Selection
The MedeA Environment provides flexible support for forcefields for use within LAMMPS. Atom type assignments are made automatically on the basis of standard template rules [10]. Several popular forcefields are supplied in the MedeA Environment for use with LAMMPS. The present investigation employs the PCFF+ forcefield, which is based on the PCFF forcefield [3] with extensions and refinements, in particular for non-bonded parameters, which have been derived as part of Materials Design’s R&D effort in a similar manner to that employed in the earlier development of the COMPASS forcefield [3], [4], [5], and [6].
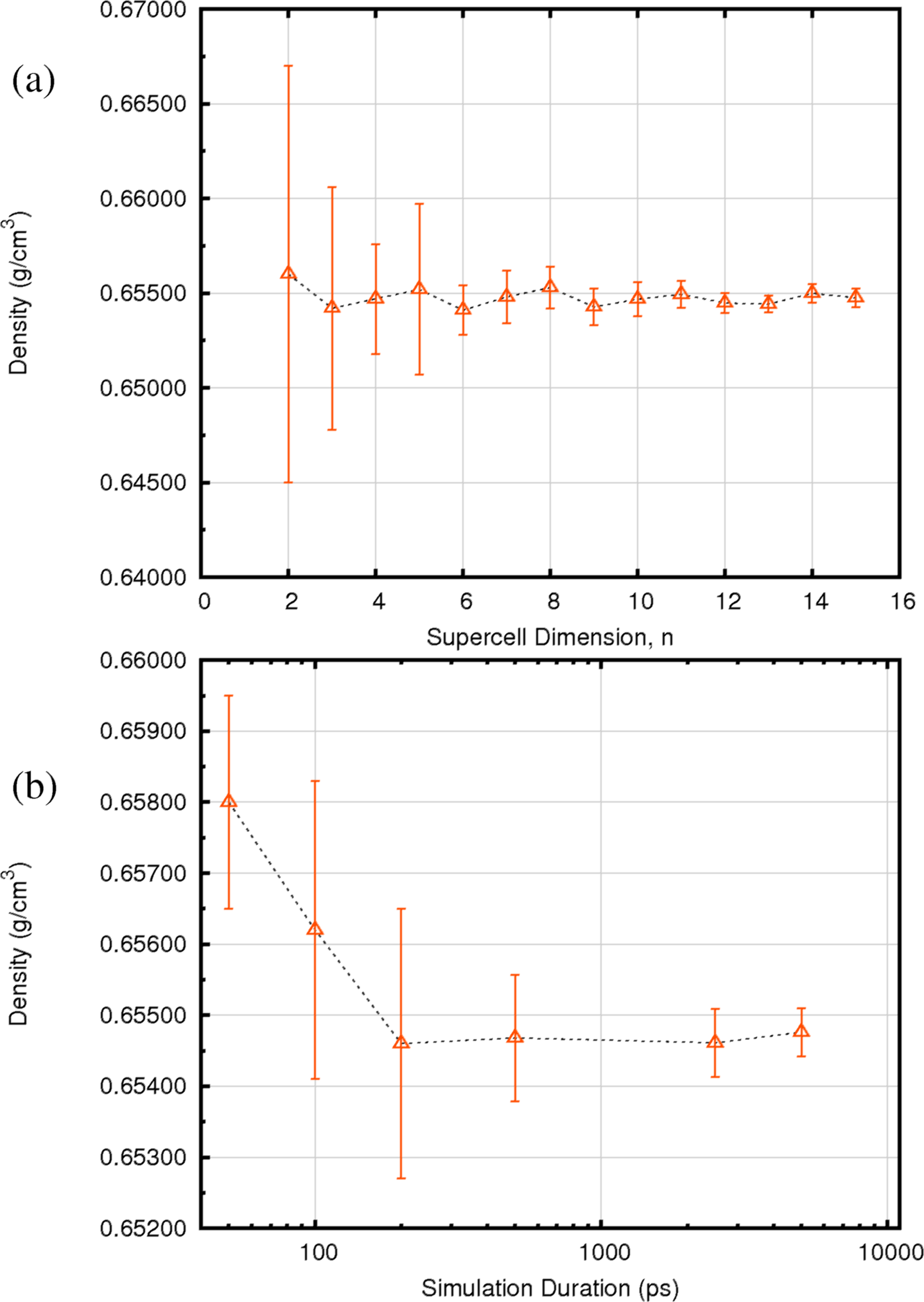
Figure 37.4.1 Variation in the calculated density of n-hexane at 25 oC and 1 atmosphere as a function of: (a) system size, (where the number of molecules in the simulation is \(n^3\)), and (b) simulation duration in picoseconds (ps). For n-hexane, converged simulation results are obtained with systems of 216 hexane molecules (i.e. where n is 6) and simulation durations of 200ps.
37.5. Simulation Strategy
The simulation strategy employed may be summarized as follows:
- Construct a hexane model employing the MedeA Polymer Builder, and NPT molecular dynamics calculations using MedeA LAMMPS.
- Assess the sensitivity of calculated properties to model size and simulation durations. Determine the optimum simulation size and duration.
- For a range of hydrocarbons from n-pentane to n-eicosane, employ the protocol developed in previous step, computing a range of properties.
37.6. Selecting Calculation Parameters
(Figure 37.4.1) shows the variation in n-hexane density, together with uncertainties estimated from the property’s fluctuations during the simulation, as a function of system size (a), and simulation duration (b). It is clear that, for hexane, models containing 216 molecules, and simulation durations of 200ps provide a reliable description of system density. Practical experience indicates that systems of 3,500 atoms and NPT trajectory durations of 200ps are more than adequate for most purposes. The experimental density of n-hexane at 298.15K is 0.6548 g/cm3 and the simulations yield a value of 0.6547 g/cm3. Hence, for n-hexane, the simulated density value (with PCFF+) is within 0.02% of the experimental value.
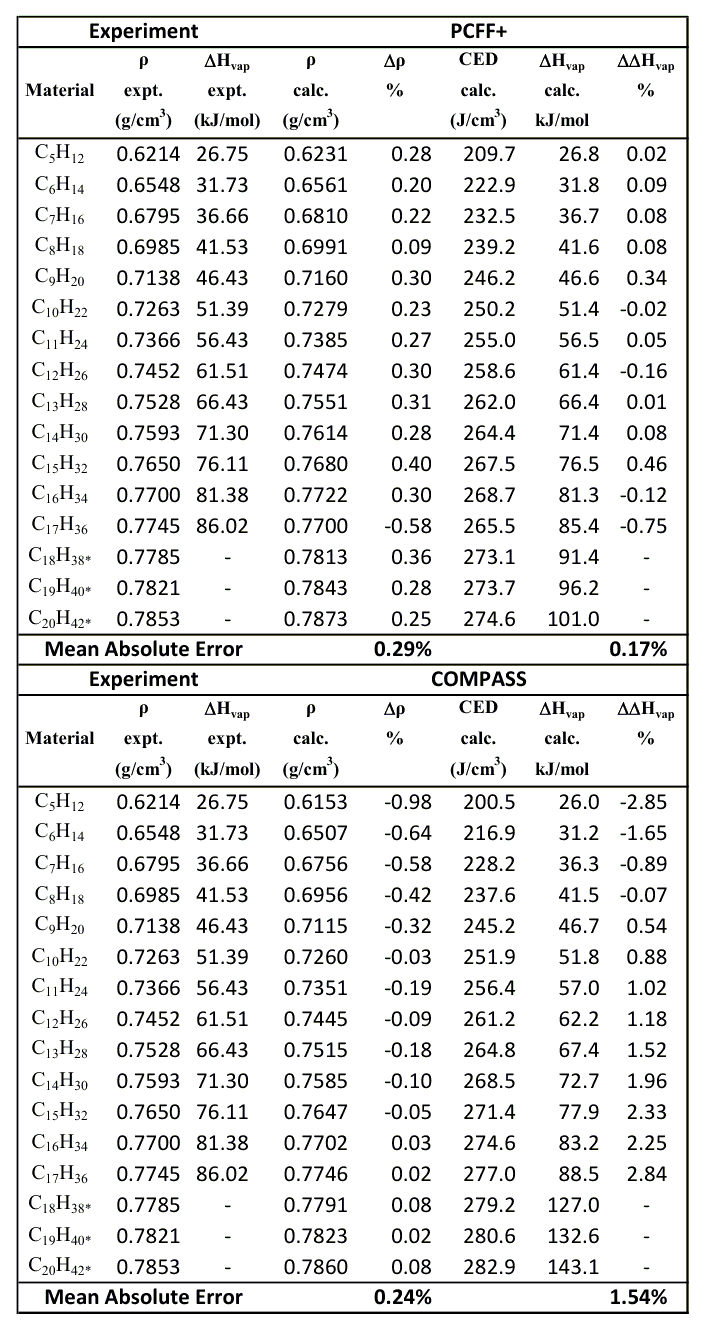
Figure 37.6.1 Computed densities (ρ), cohesive energy densities, and heats of vaporization for selected hydrocarbons at 25 oC and 1 atm. The \(\Delta\)ρ% and \(\Delta\)\(\Delta\)Hvap% values show the percentage discrepancy between experiment and calculation. Cohesive energy densities were obtained via modifications to LAMMPS made by Materials Design [11]. Experimental densities and heats of vaporization are taken from references [12] and [13] respectively. Asterisked values are estimated for the under-cooled liquid below the normal freezing point.
37.7. Computed Hydrocarbon Properties
The MedeA LAMMPS flowchart methodology enables the use or reuse of a simulation methodology with any desired system or set of systems. Hence applying this simulation protocol to a range of hydrocarbon systems is rapidly accomplished. The resulting properties are summarized in the table below. Densities, with PCFF+, are within 0.5% of experimental values. Cohesive energy densities are also tabulated. Heats of vaporization are closely related to cohesive energy density, and agreement between calculation and experiment is better than 1% (for PCFF+). Mean absolute errors are 0.29% and 0.17% for densities and heats of vaporization respectively for PCFF+. For comparison, for COMPASS, these values are 0.24% and 1.54%.
These calculated properties are based on simple atomistic models (see Figure 37.1.2) constructed using standard techniques within the MedeA Environment, atomistic molecular dynamics simulation, and the PCFF+ forcefield.
37.8. Additional Simulation Opportunities
As the tabulated results of (Figure 37.6.1) demonstrate, hydrocarbon properties can be simulated computationally with impressive agreement between calculated and experimental densities, cohesive energy densities, and heats of vaporization. It should also be emphasized that a range of additional properties may also be obtained directly from such simulation procedures. Thermal conductivity, viscosity, the variation in density as a function of pressure, for example, are also accessible, and the agreement between simulation and experiment is also, in general, excellent.
This makes computational screening in advance of experiment or even synthesis practical for many systems. Additionally, diffusive properties are similarly accessible for relatively small molecules (for example those with fewer than approximately 15 carbon atoms) and self diffusion coefficients may be simulated directly using molecular dynamics methods (see [14], for example).
There are some limits on simulated properties. Simulations are typically currently limited to trajectories extending to nano-second timescales. Hence, intrinsically slow dynamical processes, as occur in long chain polymer systems, or diffusion of large molecules, present difficulties. Additionally, model sizes are typically limited to systems measured in the thousands of atoms.
However, LAMMPS is able to exploit parallel compute resources with ease and microsecond simulation durations are becoming now common [15], and million and billion atom simulations have been reported by several groups (see [16] and [17], for example). Given the sound methodological basis of the calculations, and the increasing affordability of computational resources, it is quite clear that molecular dynamics simulations will be of ongoing importance and impact in the prediction and rationalization of the properties of organic materials
MedeA modules used in this application
- MedeA Environment
- MedeA Polymer Builder
- MedeA Amorphous Materials Builder
- MedeA LAMMPS
- MedeA JobServer
| [1] | MedeA and Materials Design are registered trademarks of Materials Design, Inc. |
| [2] | Calculations based on empirical energy functions and parameters derive from early analysis of vibrational properties of molecular systems, leading the use of the term forcefield to describe functional form and its parameterization. |
| [3] | (1, 2, 3) H. Sun, S. J. Mumby, J.R. Maple, A. T. Hagler, J. Phys. Chem. 99 , 5873 (1995) |
| [4] | (1, 2) H. Sun and D. Rigby, Spectrochimica Acta A153, 1301 (1997) |
| [5] | (1, 2) D. Rigby, H. Sun, B.E. Eichinger, Polymer International 44, 311 (1997) |
| [6] | (1, 2) Sun, H. J. Phys. Chem. B102, 7338 (1998) |
| [7] | H. Sun, P. Ren, J.R. Fried, Comput. Theor. Polymer Sci. 8, 229 (1998) |
| [8] | S. Plimpton J. Comp. Phys. 117, 1 (1995) |
| [9] | The MedeA Amorphous Materials Builder is part of the MedeA Environment. |
| [10] | Forcefield assignment template rules employ a chemical pattern language which for the recognition of specific groups of atoms and the assignment of appropriate forcefield types. Partial charge assignments are also made automatically, on the basis of bond charge increments. |
| [11] | Modification to LAMMPS for cohesive energy density calculation made by Materials Design. The source code for these changes is available upon request. The results of Table 1 were obtained with 500ps trajectories with 1000 molecules in the simulation cell. |
| [12] | F.D. Rossini, Selected Values of Physical and Thermodynamic Properties of Hydrocarbons and Related Compounds, Carnegie Press, Pittsburgh (1953) |
| [13] | Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation , Blackwell Scientific Publications, Oxford (1985) |
| [14] | Y. Iwai, H. Higashi, H. Uchida, Y. Arai, Fluid Phase Equilibria 127 , 251 (1997) |
| [15] | M.L. Klein, W. Shinoda, Science 321, 798 (2008) |
| [16] | M. Buehler, A. Hartmaier, M. Duchaineau, F.F. Abraham and H. Gao Acta Mech Sinica 21, 103 (2005) |
| [17] | F. Abraham, R. Walkup, H. Gao, M. Duchaineau, T. D. De La Rubia, M. Seager Proc. Natl. Acad. Sci. 99 , 5783 (2002) |
| download: | pdf |
|---|