38. GPU Acceleration in LAMMPS
38.1. Benchmarks of supported potentials on GPUs
We have performed benchmark calculations on a compute node equipped with dual-socket, 16-core, Intel Xeon E5-2698 @ 2.30GHz CPU (codename Haswell) and 1 NVIDIA Tesla V100 GPU card.
In the following plots, we compare the following scenarios:
Running on 8 MPI ranks (horizontal yellow lines)
Running on 32 MPI ranks (blue curves)
Running on Linux with 1 GPU card paired with 1 MPI rank (orange curves)
Running on Windows with 1 GPU card paired with 8 MPI ranks (gray curves)
The vertical axis “Speedup” is the speedup obtained when comparing to running on 8 MPI ranks. For example, if a calculation took 15 minutes on 8 cores (MPI ranks) and 5 minutes on 32 cores (MPI ranks), then the speedup is 3. The basis of comparison, speedup of 1, is provided in the plots as the yellow line. Speedup of 1 indicates the speed is the same as running on 8 MPI ranks.
38.2. LJ potential
The benchmark calculations were performed with amorphous Ar:

For larger systems (> ~500 atoms), running on 32 MPI ranks is roughly 3 times faster than running on 8 MPI ranks. On Linux, 3–25 times speedup can be observed with different sizes, while on Windows, the speedup ranged from 0.5 to 12. This means it is only faster to run on the GPU on Windows for larger sizes (> ~8,000 atoms).
38.3. EAM potential
The benchmark calculations were performed with Cu in the FCC phase:
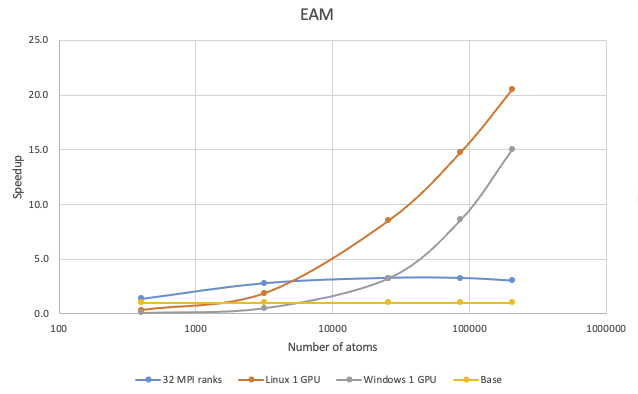
On Linux, 1 V100 GPU is faster than 8 MPI ranks and 32 MPI ranks for more than 1,500 atoms and more than 6,000 atoms, respectively. On Windows, the cross-over size over 8 MPI ranks is roughly 8,000 atoms and roughly 20,000 atoms over 32 MPI ranks. If you have a million-atom EAM simulation, running on the V100 card on Linux can be more than 30 times faster.
38.4. Tersoff potential
The benchmark calculations with the Tersoff potential were performed with Si in the diamond phase:
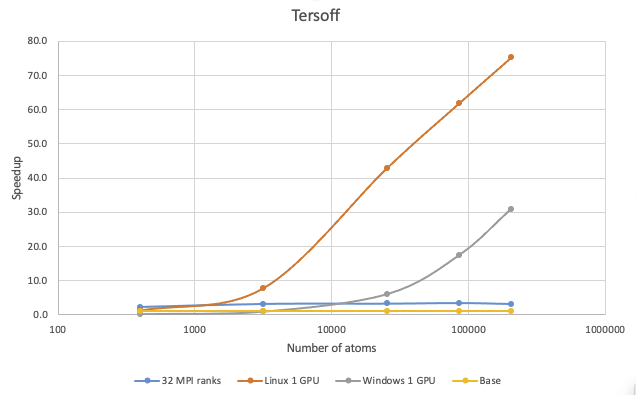
It is difficult to accelerate Tersoff simulations by just increasing the number of cores, but running on GPUs can result in significant speedup. If your Tersoff simulations have more than 100,000 atoms, running on V100 on Linux can be more than 65 times faster and roughly 20 times faster on Windows.
38.5. Buckingham potential with PPPM
The benchmark calculations with Buckingham potential were performed with SiO2 in the quartz phase. The first benchmark used the PPPM method with 10-5 precision for treating the long-range Coulombic interactions.
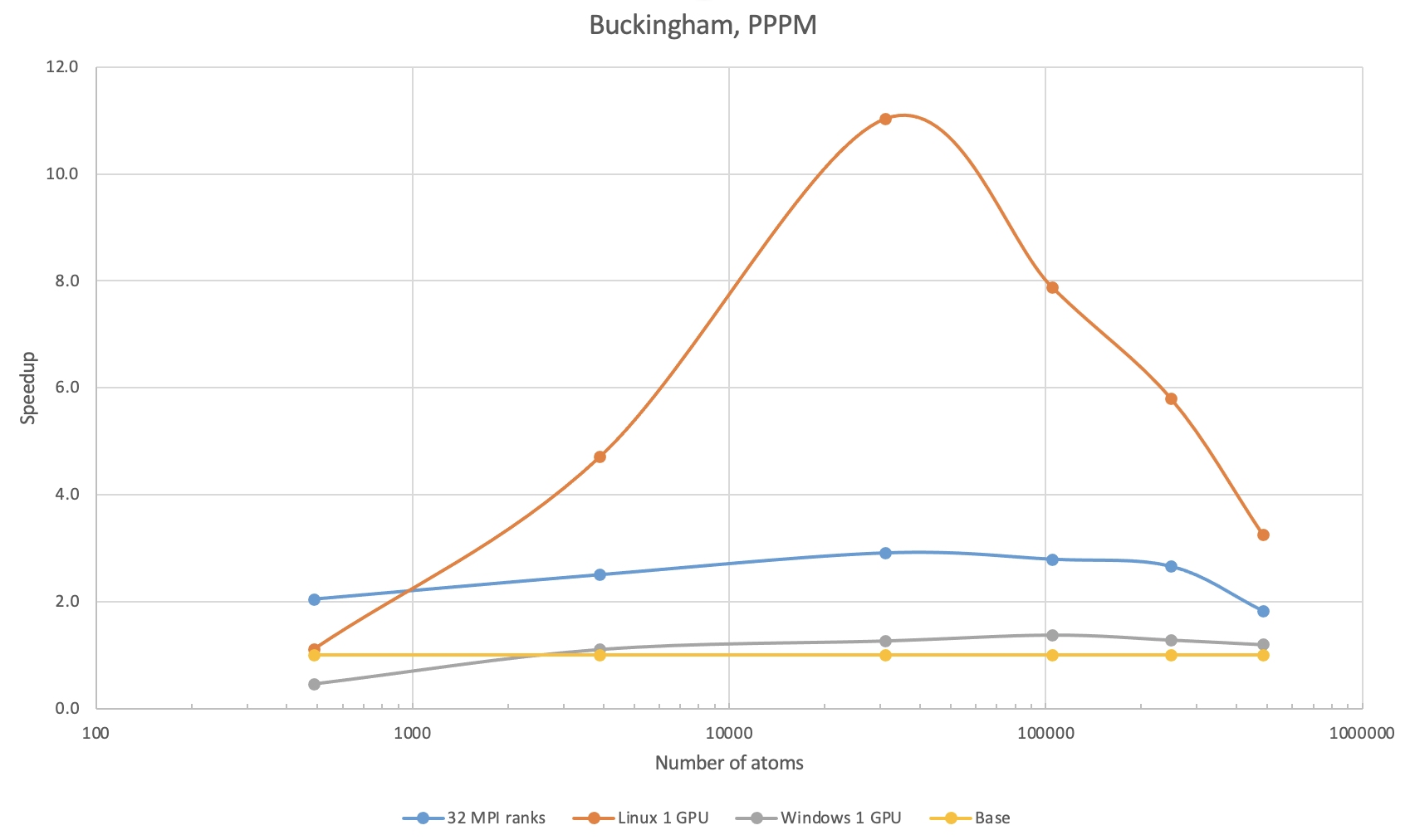
With 4 times as many MPI ranks, the speedup obtained was no larger than 3. With 1 V100 on Linux, there is an increase in speedup followed by a decrease, which is due to the PPPM method being less efficient on GPUs with increasing system sizes. With 1 V100 on Windows, the speedup was poorer than that on 32 MPI ranks, which is also due to the fact that PPPM is less efficient on the Windows implementation.
38.6. Buckingham potential with simple cutoff
The benchmark calculations with the Buckingham potential were performed with SiO2 in the quartz phase: The second benchmark used simple 9.5 \({\mathring{\mathrm{A}}}\) cutoff.
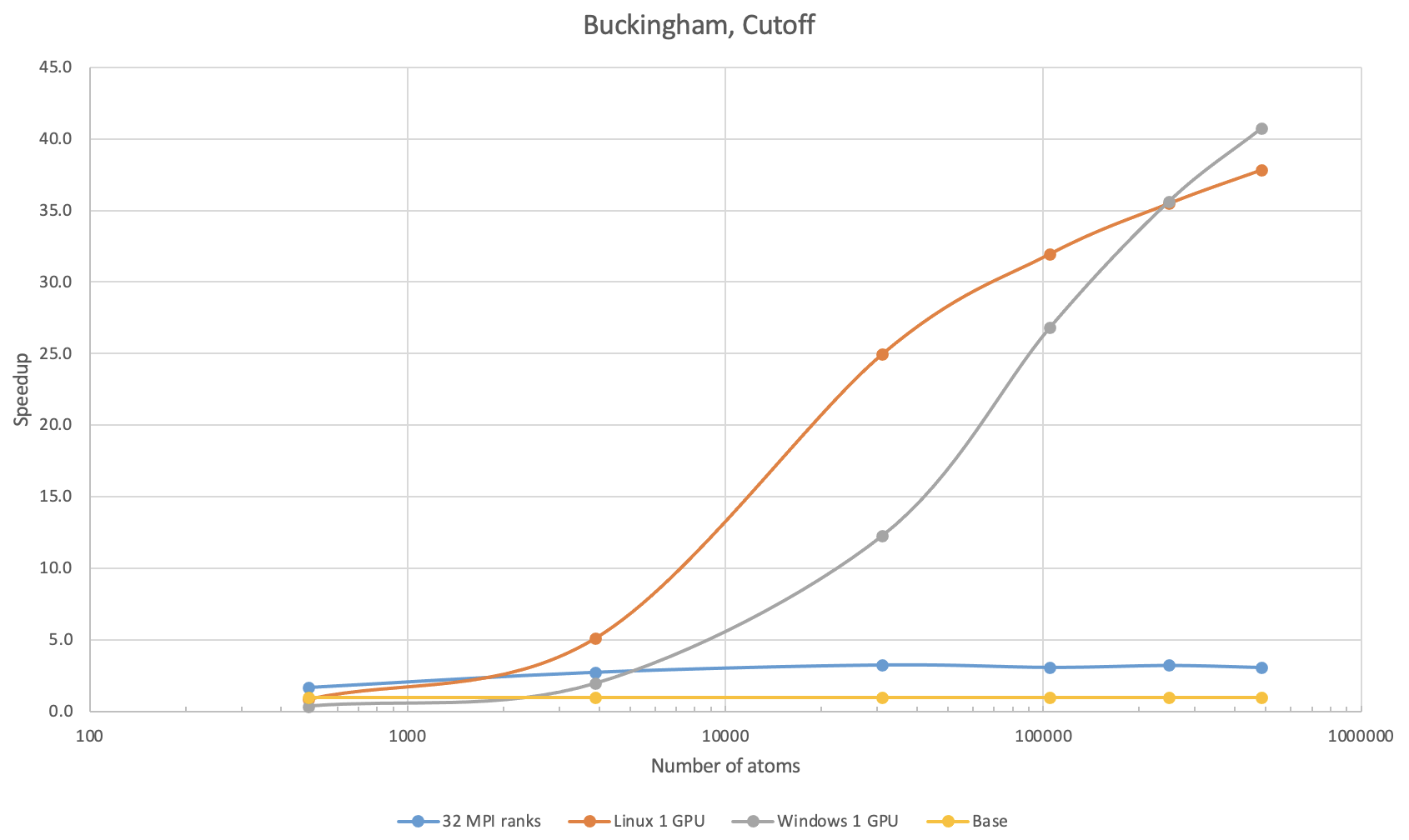
On 1 V100 on Linux, speedup over 8 MPI ranks can be observed with systems larger than 300 atoms. For 100,000 atoms, running on 1 V100 on Linux can be more than 30 times faster than on 8 MPI ranks and approximately 10 times faster over 32 MPI ranks. The Buckingham potential with a simple cutoff is accelerated well on GPUs on Windows, too. A speedup over 8 MPI ranks can be observed with more than 1,200 atoms, while that over 32 MPI ranks can be observed with systems with more than 5,000 atoms. With more than 250,000 atoms, 1 V100 on Windows is a bit more performant than that on Linux.
38.7. PCFF+ potential with PPPM
The benchmark calculations with the PCFF+ potential were performed with polystyrene. The first benchmark used the PPPM method with 10-5 precision for treating the long-range Coulombic interactions.

On 1 V100 on Linux, speedup over 8 MPI ranks can be observed with systems larger than 2,000 atoms. For 100,000 atoms, running on 1 V100 on Linux is approximately 6 times faster than on 8 MPI ranks and about 2 times faster over 32 MPI ranks. On Windows, running on the V100 card does not provide benefit over 32 MPI ranks, though it is still faster than 8 MPI ranks with more than 5,000 atoms.
38.8. PCFF+ potential with simple cutoff
The benchmark calculations with the PCFF+ potential were performed with polystyrene. The second benchmark used a simple 9.5 \({\mathring{\mathrm{A}}}\) cutoff.
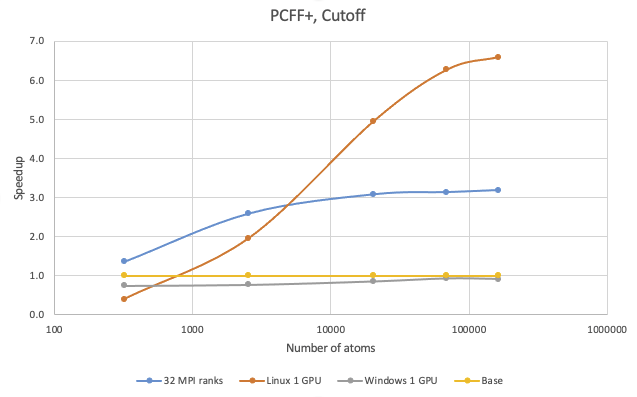
On 1 V100 on Linux, speedup over 8 MPI ranks can be observed with systems larger than 800 atoms. For 100,000 atoms, running on 1 V100 on Linux is approximately 6.5 times faster than on 8 MPI ranks and about 2 times faster over 32 MPI ranks. On Windows, running on the V100 card does not provide any benefit over 8 MPI ranks.
38.9. ReaxFF potential
The benchmark calculations with ReaxFF potential were performed with TATB in the crystalline phase:
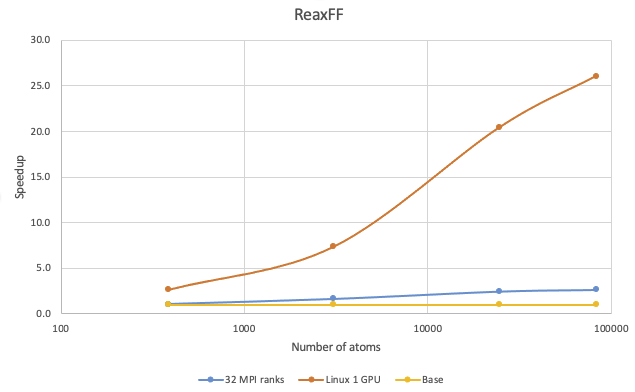
Speedup on 1 V100 on Linux over 8 MPI ranks and 32 MPI ranks can be observed with systems larger than 200 atoms. With more than 10,000 atoms, running on just 1 V100 card paired with 1 MPI rank can achieve more than 15 times the speedup over 8 MPI ranks.
Note
ReaxFF is not yet supported on GPUs on Windows.
38.10. SNAP potential
The benchmark calculations with the Tersoff potential were performed with Si in the diamond phase:
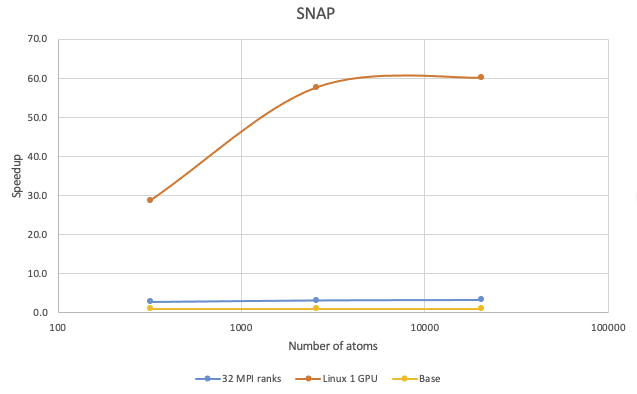
Very significant speedups can be observed with running the SNAP potential on GPUs on Linux. With 1,000 atoms the speedup can be more than 45 times, while with 10,000 atoms the speedup can be more than 60 times.
Note
SNAP is not yet supported on GPUs on Windows.
38.11. Conclusions
For best performance on Linux, please pair 1 CPU core with 1 GPU card, while on Windows, you can use as many CPU cores as appropriate.
We provided benchmarks of the Lennard-Jones, EAM, Tersoff, Buckingham, PCFF+, ReaxFF, and SNAP potentials on NVIDIA Tesla V100 GPU versus Intel Haswell CPUs. The general rule of thumb for GPU performance is: larger the system better the performance/speedup. This is because GPU cards have hundreds or thousands of cores operating at slower frequencies and the shared-memory parallelism technique becomes more efficient with increasing size. The “crossover” size - meaning the size for which GPU calculations become faster than CPU calculations - differs for each potential.
38.12. Appendix: Hardware Requirements
MedeA LAMMPS supports NVIDIA GPU cards with compute capabilities (https://developer.nvidia.com/cuda-gpus) from 6.1 to 9.0. This includes the Kepler, Maxwell, Pascal, Volta, Turing, and Ampere series GPU cards. For HPC cluster compute nodes the Tesla Kepler, Pascal, Volta, and Ampere series are recommended. For workstations and desktops, GeForce Titan V, and Quadro GP100 and GV100 are recommended. Both Linux and Windows are supported.
Though GPUs on Windows are supported, due to performance, it is strongly recommended to use/install GPUs on Linux computers.
Modules Required
MedeA Environment
MedeA EAM
MedeA ReaxFF
MedeA Machine-Learned Potential
- download: