4. Micelle Formation by a Short Chain Cationic Surfactant
The formation of micelles by surfactants was followed by molecular dynamics calculations performed with MedeA®[1] LAMMPS and using the PCFF+ forcefield. An initial model with a random distribution of C9TAC surfactant molecules was built using the MedeA building capabilities, such as the MedeA Molecular Builder and the MedeA Amorphous Materials Builder. Results are in agreement with previous simulation studies [2] and available experimental data [3].
Keywords: surfactants, self-assembly, micelle formation, colloids, n-nonyltrimethylammonium chloride, PCFF+
4.1. Surfactants
Surfactants are used as performance additives for a wide variety of industrial as well as personal and home care applications: household detergents, cosmetics, industrial cleaning, enhanced oil recovery, hydraulic fracturing, crop protection, paints and coatings, textile softeners, etc. Surfactants reduce the surface tension of a liquid, the interfacial tension between two liquids, or that between a liquid and a solid.
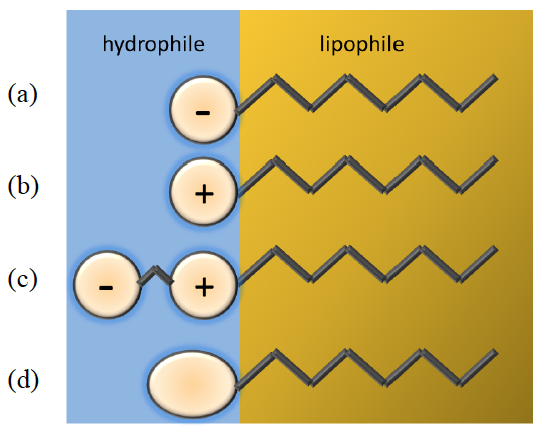
Figure 4.1.1 Classification of surfactants according to the nature of the head (a) anionic (b) cationic (c) amphoteric (d) non-ionic.
They are usually organic amphiphilic compounds consisting of a lipophilic (hydrophobic) tail and a hydrophilic head:
Tail: they can have one or two tails usually consisting of hydrocarbon chains which are fairly similar in most surfactants
Head: depending on the nature of the hydrophilic part they can be classified as (see Figure 4.1.1):
Anionic
Cationic
Amphoteric
Non-ionic
Due to the amphiphilic nature of these systems, aggregates such as micelles (Figure 4.1.2) are formed in bulk aqueous phase at intermediate surfactant concentrations. The lipophilic tails form the core of this self-assembled structure and the hydrophilic heads form an outer layer in contact with water.
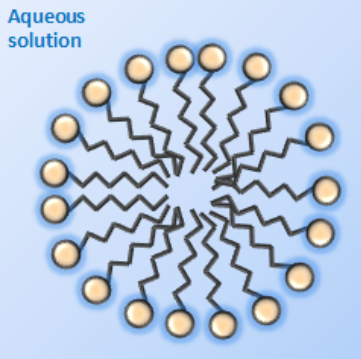
Figure 4.1.2 Surfactant micelle in water. Micelles are particles of colloidal dimensions that exists in equilibrium with the molecules or ions in solution from which it is formed.
The mechanism of micelle formation can be followed at the molecular level using molecular dynamics simulations starting from a random distribution of surfactant molecules in aqueous solution.
4.2. Molecular Modeling
Figure 4.2.1 shows a molecule of C9TAC (n-nonyltrimethylammonium chloride), a cationic surfactant with a relatively short chain (9 carbon atoms).
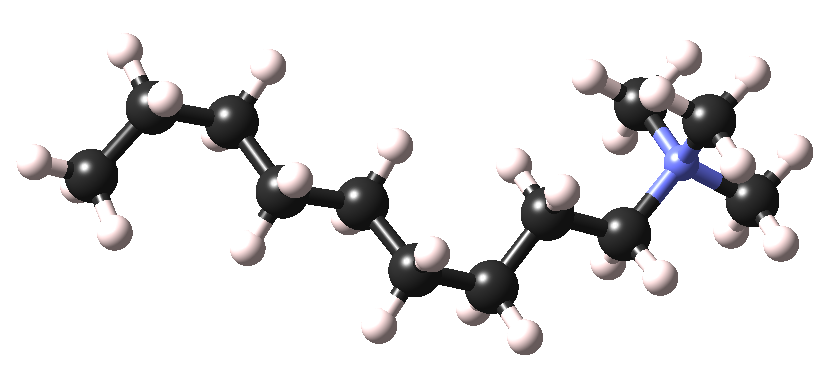
Figure 4.2.1 C9TAC surfactant molecule (only the cationic part of the molecule is displayed).
A molecule of C9TAC can easily be sketched using the MedeA Molecular Builder. Using the MedeA Amorphous Materials Builder, we generate an amorphous model containing 48 molecules of C9TAC and 3,000 molecules of H2O at a temperature of 300 K and at an initial density of 0.8 g/ml.
This constructed cell is first subjected to an energy minimization. After minimization, molecular dynamics at a constant temperature of 300 K and pressure of 1 atm (NPT-MD) takes place for a period of at least 3.5 ns. The time step is 0.5 fs for the initial 100 ps and 1 fs for the rest of the dynamics. The LAMMPS simulation program (Large-scale Atomistic-Molecular Massively Parallel Simulator ) was used for this computation. The forcefield used is PCFF+ forcefield [4].
4.3. Results
Figure 4.3.1a shows the initial random distribution of the surfactant cations at a concentration above the expected critical micelle concentration. After only 100 ps of NVT molecular dynamics run (see Figure 4.3.1b), different areas of high and low density can be seen in the model. The snapshot taken at 500 ps NVT-MD (see Figure 4.3.1c) shows the formation of three aggregates that already suggest a spherical shape. The chains, which are not part of the aggregates, are clustered in smaller groups of 2-4 molecules. Figure 4.1.1d shows the arrangement of the molecules at the end of the simulation, after 3.6 ns. Three spherical micelles can be distinguished. They contain 16, 15 and 10 molecules respectively. The rest of the surfactant molecules form a cluster with a rather indistinct shape.

Figure 4.3.1 Distribution of C9TAC cations (the water molecules and the chloride counterions are not displayed for clarity) (a) Initial random distribution generated by the MedeA Amorphous Materials Builder (b) after 100 ps NPT-MD (c) after500 ps NPT-MD (d) after 3600 ps NPT-MD.
These micelles are better visualized in Figure 4.3.2.
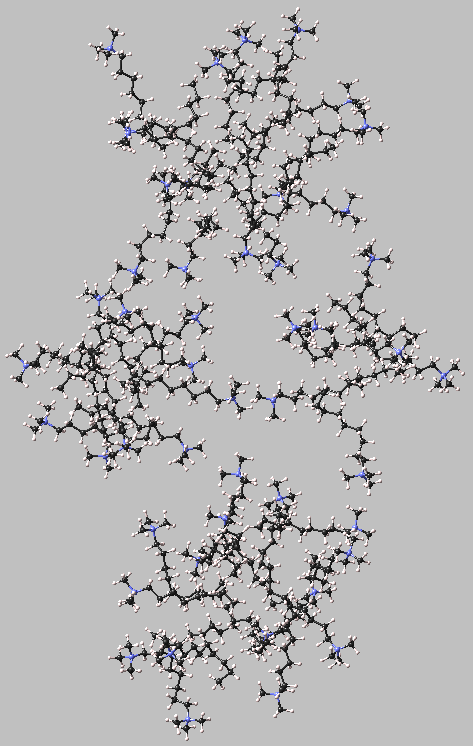
Figure 4.3.2 Distribution of C9TAC cations (the water molecules and the chloride counterions are not displayed for clarity) after 3600 ns NPT-MD.
The radial distribution function between the nitrogen atoms of the surfactant molecules averaged over the last 400 ps of simulation is shown in Figure 4.3.3. Note the main peak at around 9 \({\mathring{\mathrm{A}}}\) in agreement with previous calculations . Water molecules and chloride ions are organized to a high extent around the nitrogen atoms of the surfactant molecules. This can be observed in Figure 4.3.3 showing the radial distribution function calculated between the nitrogen atoms of the surfactant molecules and the oxygen atoms of the water molecules (red line) and between then nitrogen atoms and the chloride ions (blue line). Water molecules seem to be somewhat closer to the cationic head of the surfactant than chloride ions.
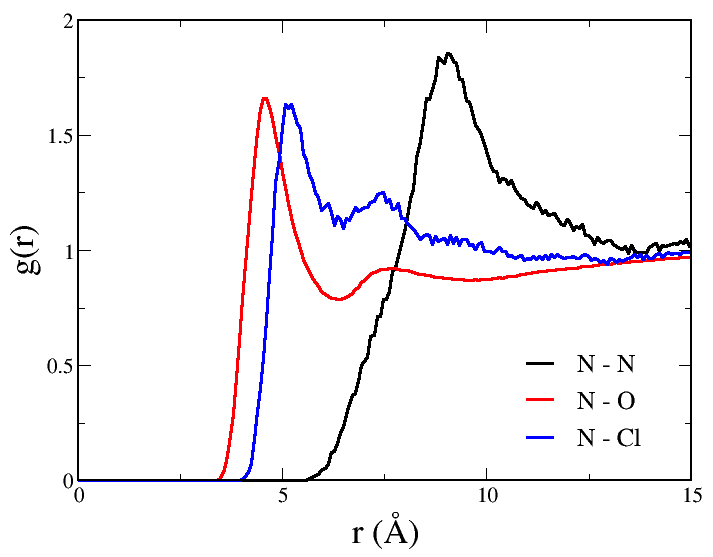
Figure 4.3.3 Radial distribution function for the nitrogen atoms of the C9TAC surfactant molecules (black line), between the nitrogen of the C9TAC molecules and the oxygen atoms of the water molecules (red line) and between the nitrogen of the C9TAC surfactant molecules and the chloride ions (blue line), averaged over the last 400 ps of NPT-MD run.
4.4. Conclusions
Atomistic, explicit-solvent simulations can provide a detailed understanding of the static and dynamic behavior of surfactant micelles. As PCFF+ is valid in a large range of conditions for numerous chemical families, MedeA is an excellent tool for exploring the influence of operating conditions (temperature, salinity, pressure, co-surfactants, etc.) for optimal formulation of additives in a given context. The MedeA flowchart interface allows the construction of automated complex workflows that can be used with a collection of different initial systems facilitating these types of studies. For example, the effect of salinity on micellization can be assessed using a single flowchart that encompasses the minimization, long molecular dynamics calculation and calculation of radial distribution functions on a set of initial models with different salt concentration.
The results shown here were obtained in less than 3 days using a standard workstation with 12 cores. C9TAC is a short chain surfactant, but the moderate computational requirements needed for this calculation indicate that this type of study can be performed for larger surfactants with very reasonable effort.
4.5. Perspectives
Molecular simulations contribute to the understanding of surfactant dynamics in simple systems as well as in mixtures with co-surfactants. Micelle formation mechanisms may be described quantitatively by following the time evolution of surfactant self-assembly.
The atomic-level understanding of surfactant dynamics and kinetics in complex environments is a key ingredient in designing high-quality additives for specialized industrial applications encountered in the oil and gas industry: from oil well drilling, hydraulic fracturing, reservoir injection, oil well production and surface plant processes to pipeline and seagoing transportation of petroleum emulsions.
- download: