MedeA Cohesive Energy Density - Compute Key Thermodynamic Characteristics of Molecular Systems
At-a-Glance
The MedeA®[1] CED module automates calculation of the cohesive energy density of molecular systems, together with the closely related solubility parameter, and heat of vaporization. Both quantities have been widely used in expressions predicting a variety of material properties, including tensile and bulk moduli, surface tension, glass temperatures, stress to initiate crazing, and thermodynamic compatibility of mixtures and blends, to cite a few examples [5].
Key Benefits
- Performs on-the-fly computation of CED, avoiding the need to save large trajectory files
- Reports van der Waals and elactrostatic components separately
- Reports related quantities such as the heat of vaporization of fluids conveniently
- Works with JobServer and TaskServer to run your calculations on the appropriate hardware, centralizing the results
- Integrates with MedeA Forcefields for advanced forcefield handling and assignment of a wide variety of organic liquids and amorphous materials
The term cohesive energy density (cohesive energy per unit volume, or CED), was introduced by physical chemist George Scatchard in his 1931 theoretical treatment of the thermodynamics of mixing of non-electrolyte solutions [2], which was the result of studies initiated more than a decade earlier by solution theory pioneer Joel Hildebrand. Here, the term “cohesive energy” represents the increase in energy of a compound if all the intermolecular forces are removed - e.g. as would occur if all molecules were to be separated by an infinite distance. Scatchard’s theory predicted that the enthalpy of mixing of a binary non-electrolyte mixture would be found by the product of the volume fractions of the components, multiplied by a term involving the differences in the square roots of the cohesive energy densities of the components. Hildebrand subsequently designated the latter as the solubility parameters (\(\delta\)i) of the individual pure components [3].
Identification of the cohesive energy with the energy required to separate molecules in a liquid by an infinite distance provides a convenient method for experimental determination of cohesive energy densities and solubility parameters from measured enthalpies of vaporization, namely using the relation:
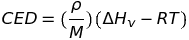
where \(\rho\) and M denote the density and molar mass, R is the gas constant, and T the temperature.
‘The MedeA CED module makes it really straightforward to include this important property calculation in any simulation workflow. It automatically performs the essential averaging over configurations without having to save large unwieldy trajectory snapshots, and delivers the results in a format easily imported into database and spreadsheet applications.’
In classical forcefield-based molecular simulations, the cohesive energy essentially corresponds to the intermolecular nonbond energy averaged over an equilibrium statistical mechanical ensemble of liquid configurations. Although this is conceptually simple, in practice the computation may require the examination of thousands of individual configurations. The CED module of the MedeA LAMMPS software is designed to perform this operation automatically without the need for post-processing of snapshots extracted from large and potentially unwieldy trajectory files. Moreover, since the nonbonded energy typically contains contributions from both coulombic and van der Waals repulsive and dispersive interactions, CED automatically reports this decomposition, which can be helpful when the solubility parameter approach is used to predict or understand thermodynamic compatibility of different materials [4]. As with other property calculations within the MedeA environment, monitoring convergence and analyzing uncertainties are performed automatically for the CED, and associated quantities are reported at the end of the simulation.
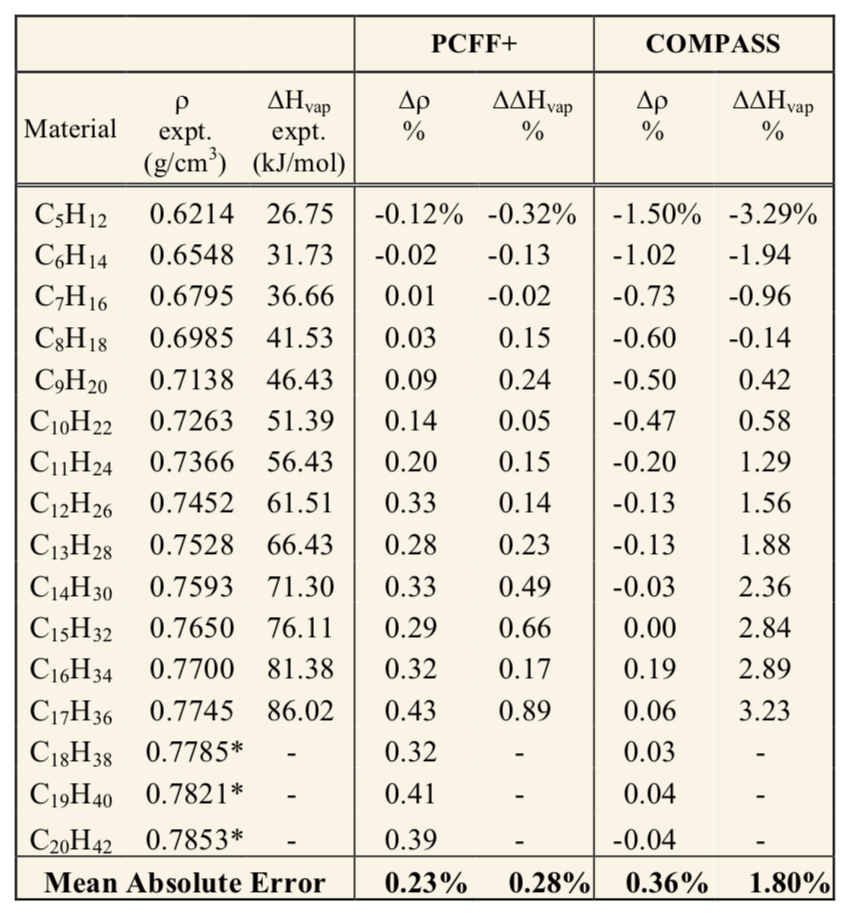
In addition to the use of the CED in correlating and predicting cohesive and adhesive properties of materials, calculation of heats of vaporization (\(\Delta\) Hv) can be particularly useful for assessing the quality of intermolecular potentials, or forcefields. This is shown in the following table, which lists CED and \(\Delta\) Hv values for a homologous series of hydrocarbons calculated using the Materials Design® PCFF+ forcefield, clearly illustrating the high accuracy achievable using MedeA software.
Computational Characteristics
- Uses the MedeA LAMMPS engine for high performance on any computer from a scalar workstation to a massively parallel cluster
Required Modules
- MedeA Environment
- MedeA LAMMPS
Find Out More
Learn more about how MedeA CED can been used to compute one of the most commonly used predictors of the properties of polymers and organic materials by viewing the following webinar:
| [1] | MedeA and Materials Design are registered trademarks of Materials Design, Inc. |
| [2] | Scatchard, G., Equilibria in Non-Electrolyte Solutions in Relation to the Vapor Pressures and Densities of the Components, Chem. Rev., 8, 321-333 (1931). |
| [3] | Hildebrand, J.H., A Critique of the Theory of Solubility of Non-Electrolytes, Chem. Rev., 44, 47-45 (1949). |
| [4] | Barton, A.F.M., CRC Handbook of Solubility Parameters and Other Cohesion Parameters, 2nd Edition, CRC Press, Boca Raton, Florida, USA (1991). |
| [5] | van Krevelen, D.W., Properties of Polymers, 3rd Ed., Elsevier, New York (1990) |
| download: | pdf |
|---|