MedeA MOPAC - Reliable High-Throughput Calculations of Thermodynamic Properties
At-a-Glance
Full integration of the quantum chemistry engine MOPAC (Molecular Orbital PACkage) within the MedeA®[1] Environment to perform more property calculations for molecules and solids of several thousands atoms faster.
Key Benefits
- Rapid and reliable thermodynamic property calculations of single molecules and crystal structures
- Accurate property predictions for organic and inorganic systems with hundreds to thousands of atoms
- Swift input generations due to excellent structure building features and structure libraries of MedeA
- Streamlined property screening for thousands of compounds
Developed and Improved over Decades
MOPAC is a semi-empirical quantum chemistry software based on the Dewar and Thiels NDDO approximation that was introduced in the 1980’s. Since then, the main developer Jimmy Stewart has constantly improved MOPAC such that properties are highly efficiently calculated with experimantal accuracy. The most recent approaches that were implemented (PM6, PM7) are as accurate as density functional (DFT) methods but significantly faster.
The impressive performance of MOPAC is especially true for thermodynamic properties of organic molecular systems as well as crystals of inorganic compounds (excluding metals).
MOPAC 2016, takes full advantage of new hardware architectures to further reduce computer time. The application range of MOPAC is very broad, comparable to DFT methods and much more versatile than approaches that use forcefields.
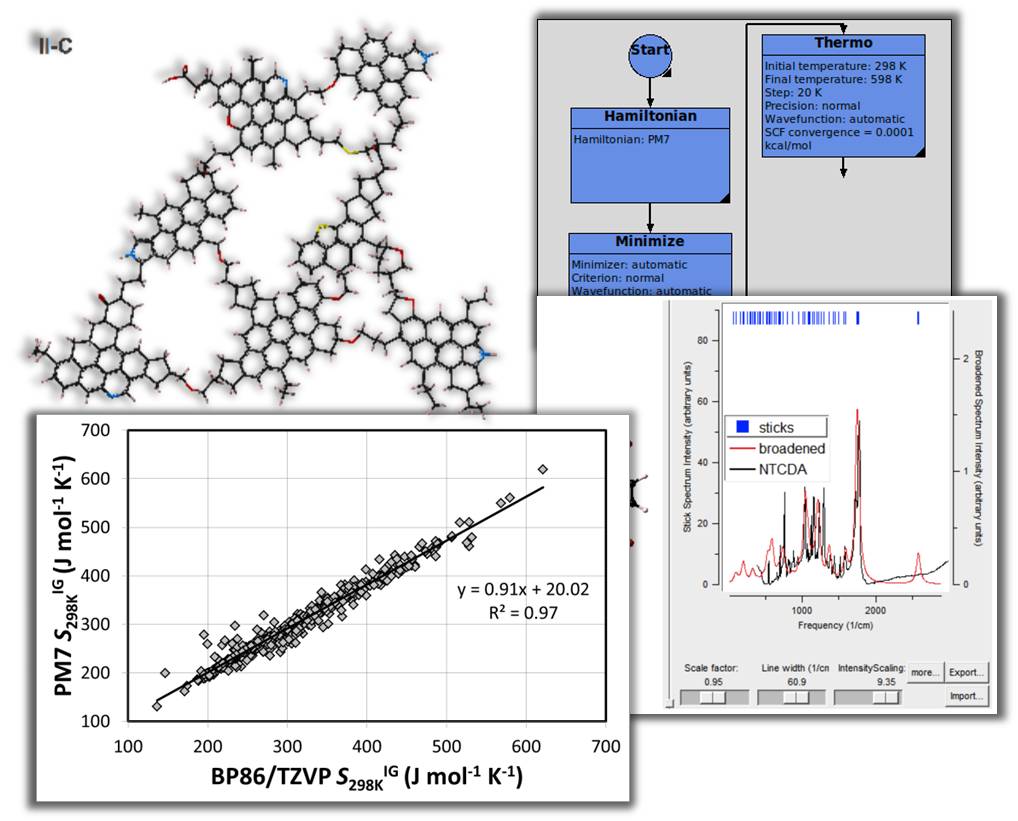
MedeA MOPAC - Calculate properties of molecules with flowcharts to compare results with experimental data.
Key Features
- Accurate predictions of properties for non-metallic systems
- More than one order of magnetide faster than standard DFT methods
- Implemented semi-empirical methods: AM1, MNDO, MNDOD, PM3, PM6, PM7, and RM1
- Combinable and compatible with MedeA LAMMPS and MedeA VASP
Why use Semiempirical methods? Modern methods have good accuracy, results can be rapidly checked, and ideas can be tested easily. We want to solve a problem!
-Jimmy Stewart
Properties
- Heat of formation
- Reaction energy and Gibbs free energy
- Solvation energy (COSMO)
- IR/Raman spectra
- UV/Vis spectra
- Molecule volume and area
- Dipole moment
- HOMO/LUMO band gap
Required MedeA Modules
- MedeA Environment
Recommended MedeA Modules
- MedeA Amorphous Materials Builder
- MedeA Docking
- MedeA GIBBS
- MedeA Gaussian GUI
- MedeA HT-Launchpad
- MedeA HT-Descriptor
- MedeA QT
Find Out More
Learn more about how MOPAC can be employed in the following Materials Design Application Notes:
- Energies of stable conformers in heavy alkanes and triglycerides using MedeA
- Properties of natural gases in classical and in HP-HT conditions
- Prediction of ideal heat capacity, Cp,id(T), of alkanolamines and amides: a combined QM-QSPR approach
- Prediction of vapor-liquid equilibrium (VLE) properties of cyclic and polycyclic compounds from Gibbs ensemble simulations
Selected Publications:
- J. J. Stewart, J. Comput. Aided Mol. Design, 4(1), 1-103 (1990)
- J. J. Stewart, J. Mol. Model., 19(1), 1-32 (2013)
- X. Rozanska, J. J. Stewart, P. Ungerer, B. Leblanc, C. Freeman, P. Saxe, & E. Wimmer, J. Chem. Eng. Data, 59(10), 3136-3143 (2014)
| [1] | MedeA and Materials Design are registered trademarks of Materials Design, Inc. |
| download: | pdf |
|---|