MedeA Analysis Tools
This is a collection of all the analysis tools that are part of MedeA.
MedeA Geometry Analysis ✓ Part of the standard MedeA Environment
The MedeA Geometry Analysis module performs interactive analysis of any crystalline or non-crystalline periodic model, providing a wealth of information including detailed symmetry analysis, atom coordinate and interatomic distance analysis, together with pair correlation functions and powder diffraction patterns.
MedeA Void Finder ✓ Part of the standard MedeA Environment
The MedeA Void Finder lets you analyze interstitial space and directly insert atoms at high symmetry points inside the chemical unit cell. It uses a Voronoi triangulation algorithm and displays candidate lattice points in a spreadsheet for analysis and processing.
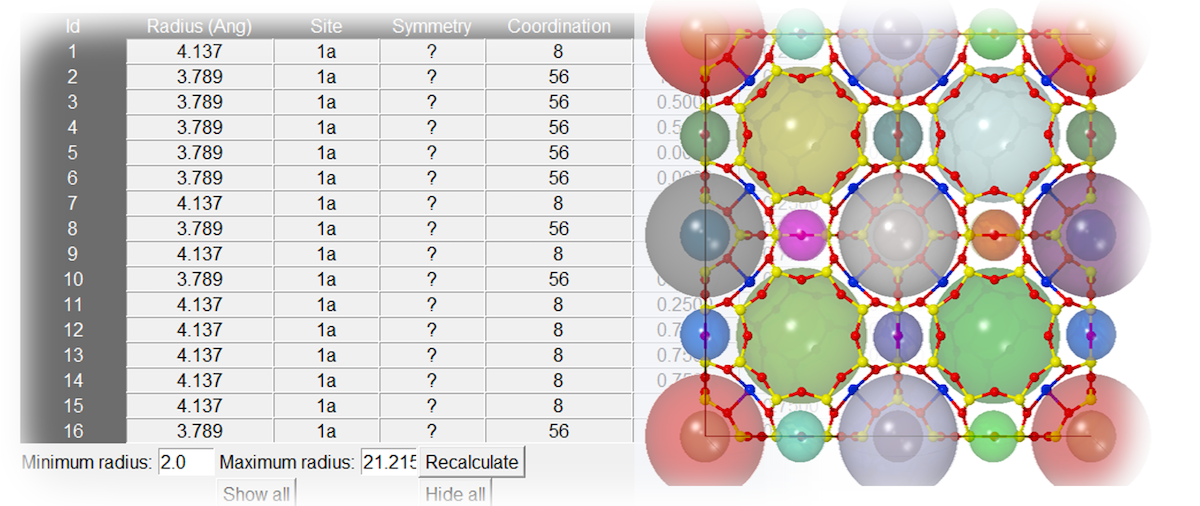
MedeA Void Finder features include:
Accounts for lattice symmetry and atomic radii
Displays interstitial sites and local coordination
Customizable upper/lower void-radius search limits
Sortable table output
One-click atom insertion and recalculation
MedeA Trajectories ✓ Part of the standard MedeA Environment
MedeA Trajectories are created during geometry optimizations and molecular dynamics simulations, monitoring the structures, energy contributions, forces and stress tensors as a function of the simulation steps. MedeA’s Trajectories can be used in forcefield optimization processes. MedeA offers visualization and animation of the structural evolution and a graph of the energy contributions.
MedeA Optical Spectra ✓ Part of the standard MedeA Environment
Optical Spectra are calculated by molecular or solid state ab initio codes in MedeA. For systems with periodic boundary conditions, frequency dependent optical functions are tensorial quantities. The MedeA Optical Spectra tool visualizes all components of the dielectric function, conductivity, refractive index, absorption index and reflectivity, and in a customized manner all combinations thereof. For molecular systems MedeA’s UV / Vis Spectra displays stick spectra and broadened spectra, including the intensities as calculated from the optical matrix elements and oscillator strengths. The predicted spectra can be overlaid by experimental data, with customized scaling of frequencies, intensities, and line widths.
MedeA Molecular Orbitals ✓ Part of the standard MedeA Environment
Analysis of MedeA Molecular Orbitals permits to qualitatively predict, describe, and understand the nature of the chemical bonds in molecules, complexes, and weakly bound clusters, the reactivity of compounds and catalysts, and the geometry of chemical species, among others.
MedeA Volumetric Rendering ✓ Part of the standard MedeA Environment
MedeA Volume Rendering offers versatile and easy to use graphical tools that allow you to visualize 3D computational data, such as total, valence, pseudo, and deformation charge densities, magnetization densities, total local potentials, electron localization functions, molecular orbitals, and Fermi surfaces. The data can be visualized as isosurfaces overlaid with the atomic structure, as color coded contour plots inside planes sliced through the 3D data set, or as planar averages along specific directions in space.
MedeA UNCLE Monte Carlo Temperature Profile
This interactive tool is used to analyze UNCLE Monte Carlo temperature profiles and to study the temperature evolution of a model. Such a study allows one to identify phase transition temperatures. Furthermore, opening Monte Carlo snapshots containing thousands of atoms from a temperature profile facilitates the study of temperature dependent structural changes within the model.
MedeA UNCLE Monte Carlo Temperature Profile tool is part of MedeA UNCLE.
MedeA Binary Ground State Diagram
Fetch the ground state diagram from a binary UNCLE cluster expansion to identify stable phases and interactively view these within MedeA.
MedeA Binary Ground State Diagram tool is part of MedeA UNCLE.
MedeA Vibrational Property Analysis
MedeA Vibrational Property Analysis tool consists of visualization tools for phonon dispersions and phonon densities of states for systems with periodic boundary conditions, as well as Infrared and Raman spectra for both molecular as well as solid state and surface systems. The phonon dispersion plots the frequencies of acoustic and optical phonon modes along a path through the Brillouin zone. Each of these modes can be animated, i.e. a structural model is created and the constituent atoms vibrate in directions and with amplitudes defined by the polarization functions. The phonon density of states displays the number of modes encountered in a certain frequency interval, with separate contributions of specific atoms and for different cartesian directions (x, y, z). From the phonon density of states accurate temperature dependent thermodynamic functions are derived. Infrared and Raman spectra including the intensities are displayed as stick spectra and broadened spectra. For molecular systems, each of the modes can be animated from the corresponding stick representation. The predicted spectra can be overlaid by experimental data, with customized scaling of frequencies, intensities, and line widths.
MedeA Vibrational Property Analysis tool is part of MedeA Phonon
MedeA Electronic Structure Analysis
MedeA Electronic Structure Analysis consists of visualization tools for band structures and densities of states. A band structure plots energy bands along a path through the Brillouin zone and allows, for instance, detailed analysis of band edge states in semiconductors and insulators. The density of states displays the number of states encountered in a certain energy interval, with separate contributions of specific atoms and for different angular momenta (s, p, d, f). These details of the density of states enables an in-depth analysis of the electronic structure and bonding.
MedeA Electronic Structure Analysis tool is part of MedeA VASP
MedeA Fermi Surface
MedeA Fermi Surface displays isosurfaces of electronic energies of metals, semiconductors, and insulators in k-space and lists the effective masses of all bands at arbitrary k-points.
MedeA Fermi Surface tool is part of MedeA Electronics
MedeA Electronic Transport
MedeA Electronic Transport provides the electronic contributions to the electric and thermal conductivity, the thermoelectric power, and related functions as based on semiclassical Boltzmann theory.
MedeA Electronic Transport tool is part of MedeA Electronics
MedeA Docking
MedeA Docking automatically creates, adjusts, and refines host-guest and surface-guest systems. The resulting host-guest structures can be employed in forcefield and first-principles based simulations.
MedeA Docking key features include:
Greatly simplifies creation of sorbate-adsorbate systems
Refines sorbate position and orientation using energy criteria
Interactive mode and integration with MedeA’s Flowcharts
Control of simulation length, Monte Carlo temperature, maximum displacements and rotation
Full integration with MedeA’s Compute Engines
MedeA Morphology
The MedeA Morphology module employs the lattice parameters, space group, and surface stabilities of a given system to determine the possible terminating faces and morphology of that crystal. These data determine the possible crystal faces and their relative orientations. The overall shape of the crystal is determined by the relative importance of each crystal face.